
Как долго живут дети с мелас синдром. Синдром мелас (melas) - прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии. Причины синдрома MELAS
Синдром MELAS (от англ. Mitochondrial Encephalomyapathy, Lactic Acidosis, and Stroke-like episodes) - митохондриалъная энцефаломиопатия, лактат-ацидоз и инсультоподобиые эпизоды - обычно манифестирует в 5-20 лет. Заболевание проявляется в первую очередь острыми инсультоподобными эпизодами с развитием очаговых изменений в затылочной и теменно-височной областях мозга и появлением соответствующей неврологической симптоматики (парезы, корковые расстройства зрения, судороги, кома, приступы головной боли и рвоты и др.). Появление очагов связывают с преходящей дисфункцией окислительного фосфорилирования в паренхиме мозга, а также структурно-метаболическими нарушениями в стенках артериол и капилляров; характерной особенностью таких «смешанных» по генсзу инфарктов мозга является относительно быстрое восстановление.
При синдроме MELAS могут наблюдаться также миопатические проявления (повышенная утомляемость и непереносимость физических нагрузок), демепция, атаксия, дегенерация сетчатки, нейросенсорная глухота, низкорослость, диабет, кардиомиопатия и целый ряд других мультиорганных проявлений. Характерен значительный уровень лактат-ацидоза в крови и спинномозговой жидкости, при биопсии скелетных мышц нередко выявляется феномен «рваных красных волокон». Синдром MELAS наследуется по материнскому типу, однако исключительная вариабельность клинических проявлений может весьма затруднять оценку семейного анамнеза.
У больных с синдромом MELAS описано, как минимум, 8 точковых мутаций в генах мтДНК, причем 5 из них локализованы в различных участках гена тРНК) . Наиболее частой мутацией является замена A->G в положении 3243 (около 80% больных), а в целом мутации указанного гена лейциновой тРНК обнаруживаются почти в 95% случаев MELAS. В редких случаях у больных MELAS описаны точковые мутации в генах других тРНК и гене СОХ Ш-субъединицы IV комплекса дыхательной цепи. Все мутации обнаруживаются в гетероплазмическом состоянии.
Синдром NARP (от англ. Neuropathy, Ataxia, Retinitis Pigmentosa) - невропатия с атаксией и пигментным ретинитом - характеризуется, в соответствии с названием, развитием прогрессирующей периферической невропатии с мышечной слабостью, мозжечковой атаксии и пигментной дегенерации сетчатки. Как и при других митохондриальных энцефаломиопатиях, клиническая картина может быть весьма вариабельной, с наличием или отсутствием у родственников ряда дополнительных симптомов (задержка психомоторного развития, эпилептические припадки, деменция). Исследования на лактат-ацидоз и другие маркеры митохондриальной дисфункции не всегда информативны.
Тип наследования болезни материнский . У всех больных с синдромом NARP обнаруживается гетероплазмическая мутация T=>G в положении 8993 (ген АТФазы 6 - субъединицы V комплекса дыхательной цепи) . Уровень гетероплазмии является решающим для характера манифестации данной мутации: при содержании мутантной мтДНК <78% заболевание может проявляться изолированными расстройствами зрения, несколько более высокий уровень мутантной мтДНК сопровождается развитием различных вариантов синдрома NARP, тогда как у больных с уровнем мутантной мтДНК 90% и более наблюдается драматически иной фенотип с быстрым фатальным исходом - болезнь Лея .
 Оглавление темы "Митохондриальная патология нервной системы":
Оглавление темы "Митохондриальная патология нервной системы":
Синдром MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke) — митохондриальная энцефаломиопатия, лактат-ацидоз и инсульт — выделен сравнительно недавно (в 1984 г.). Заболевание связано с точковой мутацией митохондриальной ДНК, которая в 90% локализуется в гене, кодирующем синтез транспортной РНК лейцина, что препятствует его включению в белки дыхательной цепи. Как и при всех митохондриальных заболеваниях, диагностика синдрома MELAS представляет трудности, обусловленные значительным разнообразием клинической картины.
Основные клинические проявления синдрома MELAS: непереносимость физических нагрузок; рецидивирующие инсультоподобные состояния. Термин «инсультоподобные», вероятно, обусловлен тем, что в большинстве описываемых случаев ведущим клиническим проявлением является головная боль с рвотой, судорогами, часто с нарушением сознания, длительностью от нескольких часов до нескольких дней.
Во время этих атак у ряда больных нередко развиваются неврологические нарушения в виде гемианопсии, гемипареза, редко в виде афазии. При проведении КТ у 60% таких пациентов выявляются очаги пониженной плотности, полиморфные судорожные приступы; при биохимическом исследовании выявляется лактат-ацидоз; начало заболевания — в 5-6-летнем возрасте; течение болезни носит прогрессирующий характер.
Приводим собственное наблюдение MELAS синдрома.
Больной А., 6 лет, поступил в клинику с жалобами на остро развившуюся слабость в правых конечностях, которая удерживалась в течение нескольких часов, головные боли. Родился от первой беременности. Беременность и роды протекали без осложнений. Раннее психомоторное развитие ребенка соответствовало возрасту. С 1 года до 2 лет отмечались аффективно-респираторные пароксизмы. С 3 лет частые ацетонемические состояния, развивающиеся на высоте головной боли.
Во время осмотра в клинике: двигательная расторможенность, неусидчивость, быстрая утомляемость при нагрузках (как умственных, так и физических). В неврологическом статусе: черепная иннервация без особенностей, мышечная гипотония, асимметрия сухожильных рефлексов (более живые справа). Парезов нет. Атаксии нет.
При первичном проведении МРТ головного мозга в левой затылочной доле обнаружена обширная зона гиперинтенсивного сигнала в режиме Т2 и гипоинтенсивность в режиме Т1 с четкими контурами, срединные структуры не смещены. Данная радиологическая картина была расценена как явление ишемического инсульта.
В течение последующего года у ребенка трижды отмечались парциальные эпилептические приступы, повторялись эпизоды приступообразной головной боли с рвотой. Получал противосудорожную терапию. Спустя год после первого лечения в клинике мальчик поступил повторно в связи с развившимся приступом генерализованных тонико-клонических судорог, который в течение часа повторился дважды.
Наросла вялость, слабость, появилась приступообразная головная боль с однократной рвотой. Повторно проведена МРТ головного мозга — в режимах Т1 иТ2 визуализировались участки измененного сигнала в теменно-затылочных отделах с обеих сторон (причем очаг слева имел меньшие размеры по сравнению с таковым при предыдущем МРТ-исследовании). Таким образом, за прошедшие после первого инсульта несколько месяцев мальчик перенес по крайней мере еще три острых нарушения мозгового кровообращения.
Метаболические нарушения заключались в значительном увеличении уровня лактата в крови до 4,2 ммоль/л (норма до 1,7 ммоль/л) и пирувата.
Комплексный анализ результатов обследования и клиники позволил установить конкретную нозологическую форму митохондриальной энцефаломиопатии — синдром MELAS, который в клинике был установлен впервые.
В последующем ребенок получал противосудорожную терапию (топамакс 5 мг/кг/сут.), лечение, направленное на стимуляцию тканевого дыхания с использованием коэнзима Q-10, янтарной кислоты, цитомака, а также актовегин и контрикал.
Приведенный случай рецидивирующих метаболических инфарктов мозга у ребенка, обусловленных синдромом MELAS, не является единственным случаем в клинике, и банк данных о подобных больных накапливается.
Синдром МЕЛАС — это митохондриальное заболевание, характеризующееся поражением мышц и ЦНС.
MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») - прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой - инсульта, диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.
История
.
Синдром MELAS был впервые описан в 1984 году Павлакисом и коллегами; десять лет спустя Павлакис и Мицио Хирано опубликовали обзор 110 случаев заболевания.
Тип наследования:
материнский
Эпидемиология :
Точная частота заболевания не известна. В литературе имеются единичные данные о частоте заболевания. На севере Финляндии частота мутации A3243G, составляет 16.3:100 000.
Патогенез :
Мутации митохондральных ДНК, контролирующих дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования — важнейшего источника энергии для метаболических процессов в клетке.
Клинические проявления
В возрасте до 40 лет пациенты с МЕЛАС поступают с клиникой транзиторной ишемической атаки, а также с эпилепсией, неоднократной рвоты, головной болью, мышечную слабость. У данных пациентов нередко клинически выявляют деменцию.
Молодой возраст и отсутствие факторов риска, характерных для инсульта, помогает задуматься о МЕЛАС.
Лабораторные данные
Лактат ацидоз — увеличения уровня лактата и пирувата.
Данные визуализации
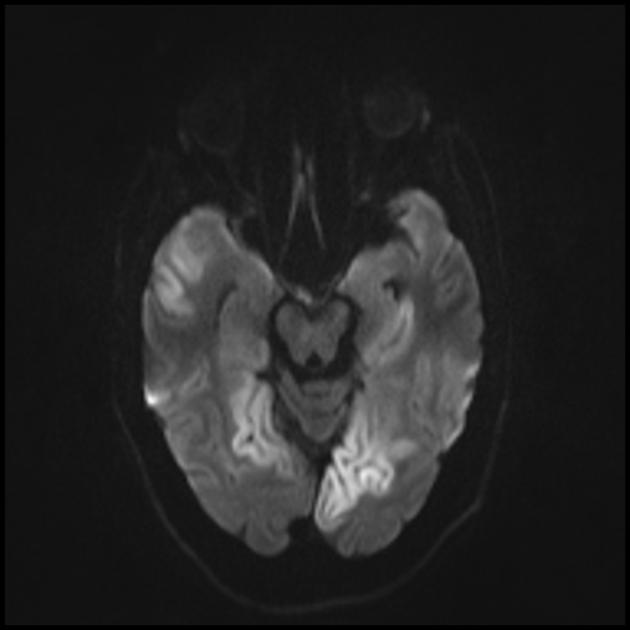
Изменения головного мозга схожи с изменениями при инсульте.
Отличия от инсульта
1) области поражения не совпадают с границами артериальных сосудистых бассейнов.
2) при повторных приступах очаги визуализируются в другой локализации.
+ клинические данные (молодой возраст, отсутствие факторов риска инсульта).
КТ
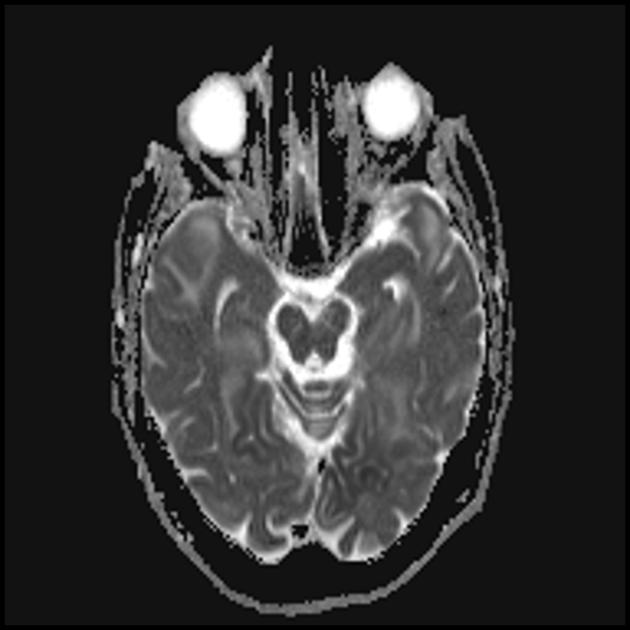
Множественные гиподенсивные области не соответствующие сосудистому бассейну.
Кальцификация базальных ганглий (наиболее чаще у пожилых пациентов).
Атрофия возникает на фоне регресса и клинического улучшения.
МРТ
Острый инфаркт
Для дифференциации с инсультом используют ADC и DWI (при инсультах ограничение диффузии (цитотоксический отек), а при МЕЛАС диффузия ограничена незначительно, либо без изменений (вазогенный отек).
Вовлечение в патологический процесс субкортикального белого вещества головного мозга.
Ухудшение визуализации четкости контуров извилин и повышение сигнала от них на Т2-взвешенных изображения.
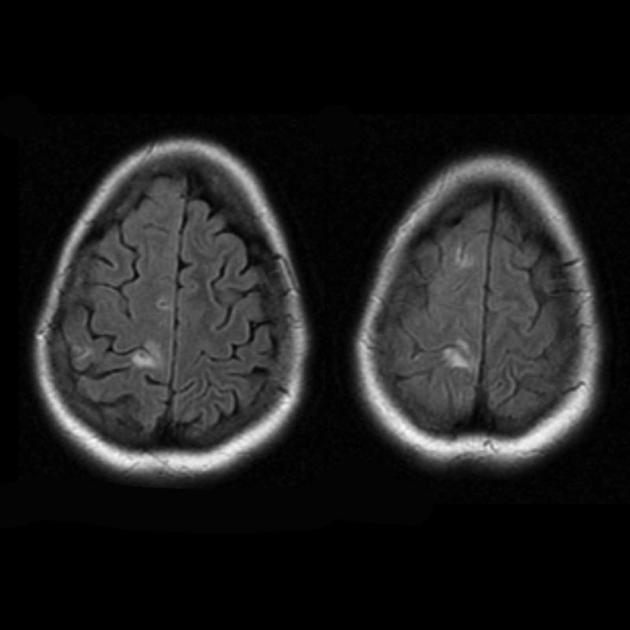
Хронический инфаркт
Изменения могут быть симметричными и ассиметричными.
Фокальная атрофия возникает на фоне регресса и клинического улучшения.
Теменная, затылочная и височная доля головного мозга наиболее чаще поражаются.
МР-спектроскопия
Повышение уровня лактата.



| Митохондриальная миопатия, encephalomyopathy, молочный ацидоз, и инсульт, как эпизоды | |
|---|---|
| Базальные ганглии кальцификация, мозжечковая атрофия, увеличение лактата ; КТ изображение человека с диагнозом MELAS | |
| Специальность | неврология |
генетика
Мышечная биопсия человека с диагнозом MELAS , но не несущая известной мутации. (а) Модифицированный Гомори трехцветный краситель показывает несколько рваных красных волокон (стрелки). (б) цитохром с оксидазы пятно, показывающий тип-1 , слегка окрашенных и тип волокна II, темные волокна и несколько волокон с аномальными коллекций митохондрий (стрелки). Обратите внимание, цитохром с оксидазы отрицательные волокна, как правило, рассматривается в митохондриальной энцефалопатии, молочнокислого ацидоза и инсульт-подобные эпизоды (MELAS). (с) Сукцинатдегидрогеназа окрашивание показывает несколько рваных синих волокон и интенсивное окрашивание в митохондриях кровеносных сосудов (стрелка). (г) электронная микроскопия показывает аномальную коллекцию митохондрий с паракристаллическими включениями (стрелок), осмиофильными включениями (большие стрелками) и митохондриальными вакуолями (малые стрелками).
МЕЛАС вызывается мутациями в генах в митохондриальной ДНК.
NADH-дегидрогеназы
Мутации в MT-TL1 причиной более 80 процентов всех случаев MELAS. Они снижают способность митохондрий, чтобы сделать белки, использовать кислород и производить энергию. Исследователи не определили, как изменения в митохондриальной ДНК приводит к специфическим признакам и симптомам MELAS. Они продолжают исследовать эффекты митохондриальных мутаций генов в различных тканях, особенно в головном мозге.
наследование
Это условие наследуются в митохондриальном узоре, который также известен как материнское наследование и гетероплазмии . Эта модель наследования относится к генам, содержащимся в митохондриальной ДНК. Из яйцеклеток, но не сперматозоиды, способствуют митохондрии для развивающегося эмбриона, только самки проходят митохондриальные условия для своих детей. Митохондриальные расстройства могут появиться в каждом поколении семьи и могут повлиять как мужчина, так и женщина, но отцы не проходят митохондриальные черты своих детей. В большинстве случаев, люди с MELAS унаследовать измененный митохондриальный ген от матери. Реже, результаты расстройство от новой мутации в гене митохондриальной и встречается у людей, не имеющих семейной истории MELAS.
диагностика
Лечение / прогноз
Пациенты управляются в соответствии с тем, что участки тела влияют на конкретный момент времени.
Вестник АГИУВ, спецвыпуск, 2013г. удк 616.8-007: 616.853.3
дебют сидромл melas с фебрильными судорогами
(случай из практики)
т. о. мусабекова, А. и. хамзина
Кыргызско-Российский Славянский Университет, Кафедра неврологии и нейрохирургии, г. Бишкек, Кыргызстан
Фебрильные судороги (ФС) известны со времен античности. Еще Гиппократ писал, что ФС наиболее часто возникают у детей первых 7 лет жизни и гораздо реже - у более старших детей и у взрослых . Но впервые термин «фебрильные судороги» применил в 1904 году B. Hochsinge для обозначения судорожных пароксизмов, развивающихся в детском возрасте на фоне лихорадки. В настоящее время предпочтительнее говорить о фебрильных приступах (ФП), а не ФС, так как в клинической картине данного состояния могут наблюдаться не только судорожные, но и бессудорожные пароксизмы 2]. По определению ILAE от 1993 года, ФП - это приступы, отмечающиеся у детей в возрасте старше 1 месяца, связанные с фебрильным заболеванием, не вызванным инфекцией ЦНС; без предшествующих судорог в неонатальном периоде и неспровоцированных приступов, а также не соответствующие критериям других острых симптоматических приступов. Согласно проекту классификации 2001 года, ФП отнесены в группу состояний, которые не требуют обязательного диагноза эпилепсии . Таким образом ФП определяются как эпизод эпилептических приступов, возникающих у детей в возрасте от 6 мес. до 5 лет при повышении температуры в период вирусного или бактериального заболевания, не связанного с нейроинфекцией и метаболическими нарушениями. Истинные ФП следует отличать от фебрильно провоцируемых приступов, которые могут входить в структуру ряда форм эпилепсии, например при синдроме Драве. В редких случаях ФП могут быть первым симптомом митохондриальных заболеваний у детей .
синдром MELAS (митохондриальная энцефало-ми-опатия с лактат-ацидозом и инсультоподобными эпизодами) был впервые выделен в отдельную нозологическую форму S. Pavlakis et al. только в 1984 г. . Заболевание относится к группе митохондриальных болезней, связанных с точковыми мутациями митохондриальной ДНК, в результате которых происходит нарушение энергопродукции в митохондриальной дыхательной цепи . Известно что точечные мутации могут возникать во многих генах (MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2), наследоваться по материнской линии . Распространённость синдрома MELAS сложно оценить из-за разнообразия проявлений и связанной с этим трудностью диагностики. к 2000 году было опубликовано более 120 наблюдений заболевания . Кардинальными симптомами при MELAS-синдроме являются: непереносимость физических нагрузок, инсультоподобные эпизоды, судороги, «рваные красные» волокна в биоптатах мышечной ткани, лактат-ацидоз и дебют заболевания в возрасте до 40 лет . Синдром MELAS следует дифференцировать с другими митохондриальными заболеваниями: синдромом Кернса-Сейера и MERRF.
Ниже приводится наше собственное наблюдение больной П. 2003-го года рождения, проживающей в городе Бишкек. Ребенок обратился к нам в центр клиники МЕБИ ЛТД города Бишкек в весной 2013 года с жалобами на тонико-клонические судороги в ногах и руках длительностью до 2 минут, протекающие с потерей сознания и развивающиеся
только на фоне повышения температуры тела выше 37 С°, а также на появление сложности в усвоении школьного материала, снижение памяти, повышенную утомляемость и мышечную слабость, неловкость при ходьбе.
Дебют заболевания у девочки отмечался в возрасте 6-ти месяцев с генерализованного тонико-клонического приступа продолжительностью до 1 минуты на фоне повышения температуры тела до 38 С°, после которого была госпитализирована в Республиканскую инфекционную больницу города Бишкек, где исключили нейроинфекцию. В последующем ФП возникали каждый раз при повышении температуры тела выше 37 С°. В возрасте 1 год при обращении в Национальный центр педиатрии и детской хирургии КР, было проведено МРТ головного мозга, ЭЭГ, где патологии не выявлено, был назначен депакина в дозе 20 мг/кг/сут. Тем не менее ФП продолжались на фоне приема противосудорожного препарата. В возрасте 5-ти лет самостоятельно обратились в Республиканскую детскуюклиническую больницу (РДКБ) города Москвы, где было повторно проведено МРТ головного мозга и видео - ЭЭГ -мониторинг (ВЭМ) дневного сна, где вновь патологии не выявлено. Врачами РДКБ был выставлен диагноз криптогенная эпилепсия, рекомендовано повысить дозу депакина до 25 мг/кг/сут. Первые три класса в общеобразовательной школе закончила на оценки «4» и «5». С 9-ти лет мама стала отмечать постепенное нарастание у ребенка быстрой утомляемости после физических нагрузок, появление сложности в усвоении школьного материала, встал вопрос о переводе ребенка в специализированное школьное учреждение для детей с умственной отсталостью. ФП продолжали беспокоить ребенка и после 7-ми лет на фоне приема депакин хроно в дозе 25 мг/кг/сут.
Из анамнеза жизни: ребенок от первой беременности протекавшей на фоне легкого токсикоза в первом триместре, юыл эпизод ОРВИ без температуры в сроке 5 мес. Роды в срок, самостоятельные в головном предлежании, по шкале Апгар 7/8 баллов, ВПР - 3340 гр., рост - 52 см. Раннее развитие ребенка соответствовало возрастной норме.
На момент осмотра в в клинике числа возрасте 10-ти лет со стороны черепно-мозговых нервов отмечается незначительная девиация языка вправо, миопатический синдром в руках и ногах в виде гипотонии, легкой гипотрофии проксимальных отделов рук и ног со снижением мышечной силы до 4 баллов, снижении сухожильных рефлексов, а также легкое пошатывание в позе Ромберга и неловкость при выполнении пальце-носовой и коленно-пяточной проб, снижение кратковременной памяти и внимания.
Дополнительные обследования: Нв 112 г/л, эритроциты 3,5 *10"12/л, печеночные тесты, общий белок, сахар, креатинин в пределах нормы.
Продолжение ФП после 5-ти лет с развитием резистентности к вальпроатам, присоединение миопатического синдрома и снижение когнитивных функций позволило высказать предположение о возможности наличия у больной митохондриальной патологии, а именно синдрома MELAS, что потребова-
ло проведения ряда дополнительных исследований. При электронейромиографии проведенной в НЦП и ДХ выявлен первично-мышечный тип поражения в виде уменьшения длительности потенциала двигательных единиц на 30-35% и снижения их амплитуды с нормальной скоростью проведения по периферическим нервам. При повтороном ВЭМ патологии не выявлено. В SVS лаборатории имени В.М. Савинова города Алматы была определена концентрация депакина в крови до приема препарата- 86, 98 нг/мл и через 2 часа после приема препарата -113, 61 нг/мл при норме 50-100 нг/мл. В Приват клинике города Алматы было определено содержание молочной кислоты натощак - 3,1 ммоль/л, при норме до 1,7ммоль/л. Был выставлен предварительный диагноз синдром MELAS, депакин заменен на кеппру, введен коэнзим Q10, карнитин, витамины группы В, витамин Е, диета с ограничением приема углеводов, рекомендовано проведение генетического обследования.
Таким образом, в представленном нами клиническом случае у ребенка наблюдались простые ФП, для профилактического лечения которых был назначен депакин с длительным приемом на протяжении нескольких лет несмотря на наличие фармакорезистентности. Принимая во внимание отсутствие четких доказательств эффективности профилактического применения антиконвульсантов у детей с ФП, назначение депакина в данном случае было нецелесообразно. Так по литературным данным длительное применение депакина и барбитуратов существенно усугубляет течение митохондриальных заболеваний, порой приводя к прогрессированию патологического процесса , что и произошло в нашем клиническом случае.
список литературы:
1. Гузева В.И. Специальные синдромы (ситуационно-обусловленные приступы) / В.И. Гузева // Эпилепсия и неэпилептические пароксизмальные состояния у детей.-М.: МИА, 2007.- С. 443- 457
2. Мухин К.Ю. Фебрильные судороги /А.С. Петрухин
// Неврология детского возраста. - М.: Медицина, 2004.-С.664-668.
3. Никанорова М.Ю., Темин П.А., Кобринский Б.А. Фебрильные судороги / П.А. Темина, М.Ю. Никаноровой // Эпилепсия и судорожные синдромы у детей.- М.: Медицина, 1999.- С. 169- 195.
4. Основные методы лечения детей, страдающих митохондриальными заболеваниями: Методич. указания.-М., 2001.
5. Темин П.А. и др. //Неврол. журн.- 1998. № 2.- С. 43.
6. Яхно Н.Н. и др. //Неврол. журн. 1998.- № 5.- С. 14.
7. Ban S. et al. //Acta Pathol. Jpn.- 1992. -Vol. 42. -P. 818.
8. Hirano M., Pavlakis S.G. // J. Clin. Neurol. -1994.-Vol. 9. -P. 4.
9. ILAE Commission report: glossary of descriptive terminology for ictal semiology: report of the ILAE Task Force on Classification and Terminology /Epilepsia.- 2001.- Vol. 42. -P.1212-1218.
10. ILAE. Guidelines for epidemiologic studies on epilepsy /Epilepsia.- 1993.- Vol.34.- P. 592-596.
11. Pavlakis S.G. et al. //Ann. Neurol.-1984. -Vol. 16.-P. 481.
12. Sciacco M. et al. // J. Neurol. -2001. -V 248. -P. 778.
Фебрильные судороги нередко могут быть первым симптомом митохондриальных заболеваний у детей, что значительно затрудняет своевременную диагностику и начало патогенетического лечения болезни, а порой обуславливает применение препаратов ухудшающих течение и прогноз заболевания.
ключевые слова: судороги, терапия.
Febrile seizures can often be the first symptom of mitochondrial disease in children, which greatly complicates the timely diagnosis and early treatment, and sometimes causes the use of drugs worsen the course and prognosis of the disease.
Keywords: convulsions, therapy.
удк 616.831-005.4
СЛУЧАИ ИШЕМИЧЕСКОГО ИНСУЛЬТА КАК ПРОЯВЛЕНИЯ МИТОХОНДРИАЛЬНОЙ ЭНЦЕФАЛОПАТИИ У БОЛЬНОГО МОЛОДОГО
ВОЗРАСТА
Карбозова К.З., Луценко И. Л.
Кафедра неврологии с курсом медицинской генетики, Кыргызская Государственная Медицинская Академия имени И.К. Ахунбаева,
г. Бишкек, Кыргызстан
Распространенность инсульта в молодом возрасте (до 45 лет) составляет от 2,5 до 10% всех случаев нарушений мозгового кровообращения и продолжает увеличиваться . У пациентов молодого возраста наиболее частыми причинами развития ишемических сосудистых нарушений являются: аномалии цереброваскулярной системы, диссекция, кардиальная патология мигрень, дефекты коагуляции, АФЛС, .
За 5 последних месяцев в отделении неврологии №1 Национального госпиталя при Министерстве Здравоохранения Кыргызской Республики (НГМЗКР) получало лечение 608 больных. Произведен анализ 46 (7.5) историй болезни пациентов, перенесших ишемический инсульт, из них 4 (8.7%) молодого возраста (до 45 лет по ВОЗ). В таблице 1 приведены подтипы ишемического инсульта.
Представляем историю болезни пациента, госпитализированного с первоначальным диагнозом острое нарушение мозгового кровообращения (оНмК), у которого истинную природу заболевания удалось установить только при динамическом наблюдении и специальном дополнительном обследовании.
Таблица 1.
Подтип инсульта Число больных в %
Атеротромботиче ский 32 69,5
Лакунарный 6 13,04
Гемореологический 3 6,5
Кардио эмболиче ский 3 6,5
Митохондриальный 1 2,2