
Jak długo żyją dzieci z zespołem Melas? Zespół Melas (melas) to postępująca choroba neurodegeneracyjna charakteryzująca się objawami wymienionymi w tytule. Przyczyny zespołu MELAS
Zespół MELAS(z angielskiego. Mitochondrialna encefalomyapatia, kwasica mleczanowa i epizody udaropodobne) - encefalomiopatia mitochondrialna, kwasica mleczanowa i epizody udaropodobne - zwykle pojawiają się po 5-20 latach. Choroba objawia się przede wszystkim ostrymi epizodami udaropodobnymi z rozwojem zmian ogniskowych w okolicy potylicznej i ciemieniowo-skroniowej mózgu oraz pojawieniem się odpowiednich objawów neurologicznych (niedowład, korowe zaburzenia widzenia, drgawki, śpiączka, napady bólu głowy i wymioty itp.). Pojawienie się ognisk wiąże się z przemijającą dysfunkcją fosforylacji oksydacyjnej w miąższu mózgu, a także zaburzeniami strukturalnymi i metabolicznymi w ścianach tętniczek i naczyń włosowatych; charakterystyczna cecha takie „mieszane” w genszu zawały mózgu to stosunkowo szybka rekonwalescencja.
W przypadku zespołu MELAS Można również zaobserwować objawy miopatyczne (zmęczenie i nietolerancja wysiłku), otępienie, ataksję, zwyrodnienie siatkówki, głuchotę czuciowo-nerwową, niskorosłość, cukrzycę, kardiomiopatię i szereg innych objawów wielonarządowych. Charakterystyczny jest znaczny poziom kwasicy mleczanowej we krwi i płynie mózgowo-rdzeniowym, przy biopsji mięśni szkieletowych często ujawnia się zjawisko „rozerwania czerwonych włókien”. MELAS jest dziedziczony po matce, ale ekstremalna zmienność objawy kliniczne może bardzo utrudnić ocenę historii rodziny.
U pacjentów z zespołem MELAS opisał co najmniej 8 punktowych mutacji w genach mtDNA, przy czym 5 z nich było zlokalizowanych w różnych regionach genu tRNA). Najczęstszą mutacją jest substytucja A->G w pozycji 3243 (około 80% pacjentów) i ogólnie mutacje tego genu leucyny tRNA stwierdza się w prawie 95% przypadków MELAS. W rzadkich przypadkach u pacjentów z MELAS opisano mutacje punktowe w genach innych tRNA i genie COX podjednostki III kompleksu IV łańcucha oddechowego. Wszystkie mutacje znajdują się w stanie heteroplazmatycznym.
Zespół NARP(z angielskiego Neuropathy, Ataxia, Retinitis Pigmentosa) – neuropatia z ataksją i barwnikowym zwyrodnieniem siatkówki – charakteryzuje się, zgodnie z nazwą, rozwojem postępującej neuropatii obwodowej z słabe mięśnie, ataksja móżdżkowa i barwnikowe zwyrodnienie siatkówki. Podobnie jak w przypadku innych encefalomiopatii mitochondrialnych, obraz kliniczny może być bardzo zmienny, z obecnością lub brakiem szeregu dodatkowych objawów u krewnych (opóźniony rozwój psychomotoryczny, napady padaczkowe, otępienie). Badania nad kwasicą mleczanową i innymi markerami dysfunkcji mitochondriów nie zawsze są pouczające.
Rodzaj dziedziczenia choroby macierzyński. Wszyscy pacjenci z zespołem NARP mają heteroplazmatyczną mutację T=>G w pozycji 8993 (gen ATPazy 6 - podjednostka V kompleksu łańcucha oddechowego). Poziom heteroplazmii decyduje o charakterze manifestacji tej mutacji: kiedy zawartość zmutowanego mtDNA<78% заболевание может проявляться изолированными расстройствами зрения, несколько более высокий уровень мутантной мтДНК сопровождается развитием различных вариантов синдрома NARP, тогда как у больных с уровнем мутантной мтДНК 90% и более наблюдается драматически иной фенотип с быстрым фатальным исходом - болезнь Лея .
 Spis treści tematu „Patologia mitochondrialna układu nerwowego”:
Spis treści tematu „Patologia mitochondrialna układu nerwowego”: Zespół MELAS (encefalomiopatia mitochondrialna, kwasica mleczanowa i udar) - encefalomiopatia mitochondrialna, kwasica mleczanowa i udar mózgu - został wyizolowany stosunkowo niedawno (w 1984 r.). Chorobie towarzyszy mutacja punktowa w mitochondrialnym DNA, która w 90% jest zlokalizowana w genie kodującym syntezę transferowego RNA leucyny, co uniemożliwia jego włączenie do białek łańcucha oddechowego. Podobnie jak w przypadku wszystkich chorób mitochondrialnych, rozpoznanie zespołu MELAS nastręcza trudności ze względu na znaczne zróżnicowanie obrazu klinicznego.
Główne objawy kliniczne zespołu MELAS to: nietolerancja wysiłku; nawracające stany przypominające udar. Określenie „podobny do udaru” prawdopodobnie wynika z faktu, że w większości opisywanych przypadków główną manifestacją kliniczną są bóle głowy z wymiotami, drgawki, często z zaburzeniami świadomości, trwające od kilku godzin do kilku dni.
Podczas tych ataków u wielu pacjentów często rozwijają się zaburzenia neurologiczne w postaci hemianopsji, niedowładu połowiczego, a rzadko w postaci afazji. Podczas przeprowadzania CT 60% z tych pacjentów ujawniło ogniska o niskiej gęstości, polimorficzne napady drgawkowe; badanie biochemiczne ujawnia kwasicę mleczanową; początek choroby ma miejsce w wieku 5-6 lat; przebieg choroby postępuje.
Oto nasza własna obserwacja zespołu MELAS.
Pacjent A., lat 6, został przyjęty do poradni z dolegliwościami ostrego osłabienia kończyn prawych utrzymujące się od kilku godzin, bólami głowy. Urodzony od pierwszej ciąży. Ciąża i poród przebiegały bez komplikacji. Wczesny rozwój psychomotoryczny dziecka odpowiadał wiekowi. Od 1 roku do 2 lat notowano napady afektywno-oddechowe. Od 3 roku życia częste stany acetonemiczne rozwijające się na wysokości bólu głowy.
Podczas badania w gabinecie: odhamowanie ruchowe, niepokój, zmęczenie podczas stresu (zarówno psychicznego, jak i fizycznego). W stanie neurologicznym: unerwienie czaszki bez cech, niedociśnienie mięśniowe, asymetria odruchów ścięgnistych (bardziej ożywione po prawej stronie). Nie ma niedowładu. Nie ma ataksji.
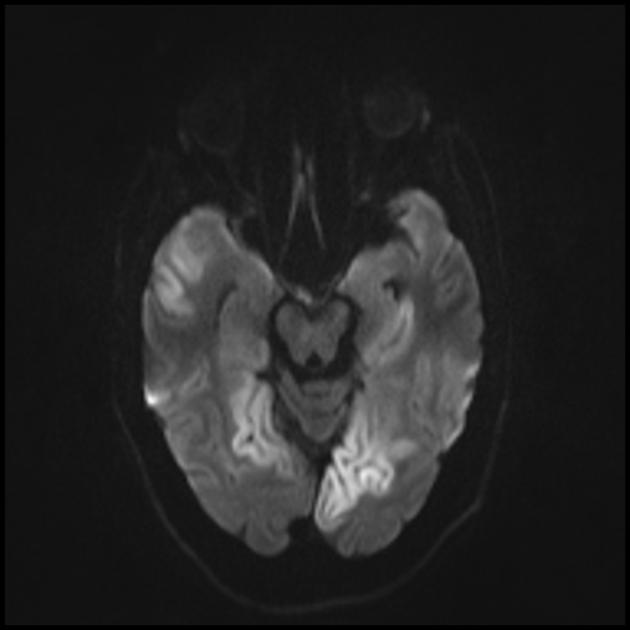
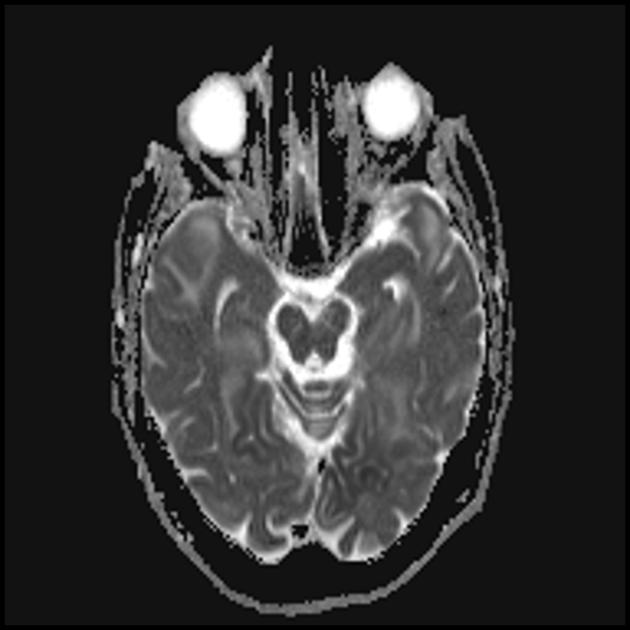
Podczas wstępnego rezonansu magnetycznego mózgu w lewym płacie potylicznym stwierdzono rozległą strefę hiperintensywnego sygnału w trybie T2 i hipointensywności w trybie T1 z wyraźnymi konturami, struktury środkowe nie uległy przemieszczeniu. Ten obraz radiologiczny uznano za zjawisko udaru niedokrwiennego.
W ciągu następnego roku dziecko miało trzy napady padaczkowe częściowe, powtarzające się epizody napadowego bólu głowy z wymiotami. Otrzymał terapię przeciwdrgawkową. Rok po pierwszym zabiegu w klinice chłopiec został ponownie przyjęty z powodu rozwiniętego napadu uogólnionych napadów toniczno-klonicznych, które powtarzały się dwukrotnie w ciągu godziny.
Zwiększony letarg, osłabienie, wystąpił napadowy ból głowy z pojedynczymi wymiotami. Powtórzono rezonans magnetyczny mózgu — w trybach T1 i T2 obszary zmienionego sygnału uwidoczniono w rejonach ciemieniowo-potylicznych po obu stronach (ponadto skupienie po lewej stronie było mniejsze niż w poprzednim badaniu MRI). Tak więc w ciągu kilku miesięcy od pierwszego udaru chłopiec doznał jeszcze co najmniej trzech ostrych incydentów naczyniowo-mózgowych.
Zaburzenia metaboliczne polegały na znacznym wzroście poziomu mleczanów we krwi do 4,2 mmol/l (norma do 1,7 mmol/l) oraz pirogronianu.
Kompleksowa analiza wyników badania i kliniki pozwoliła na ustalenie specyficznej postaci nozologicznej encefalomiopatii mitochondrialnej - zespołu MELAS, który został zdiagnozowany w klinice po raz pierwszy.
Następnie dziecko otrzymało terapię przeciwdrgawkową (Topamax 5 mg/kg/dzień), leczenie mające na celu stymulację oddychania tkankowego koenzymem Q-10, kwasem bursztynowym, cytomakiem, a także aktovegin i contrical.
Powyższy przypadek nawracających zawałów metabolicznych mózgu u dziecka spowodowanych zespołem MELAS nie jest jedynym przypadkiem w klinice, a baza takich pacjentów jest gromadzona.
Zespół MELAS to zaburzenie mitochondrialne charakteryzujące się zajęciem mięśni i OUN.
MELAS (ang. Encefalomiopatia mitochondrialna, kwasica mleczanowa i epizody udaropodobne - „encefalomiopatia mitochondrialna, kwasica mleczanowa, epizody podobne do udaru”) to postępująca choroba neurodegeneracyjna charakteryzująca się wymienionymi w tytule objawami, której towarzyszą objawy polimorficzne - udar, cukrzyca, drgawki, zmniejszenie utraty słuchu, choroby serca, niski wzrost, endokrynopatie, nietolerancja wysiłku i zaburzenia neuropsychiatryczne.
Fabuła.
Zespół MELAS został po raz pierwszy opisany w 1984 roku przez Pavlakisa i współpracowników; dziesięć lat później Pavlakis i Mizio Hirano opublikowali przegląd 110 spraw.
typ dziedziczenia:
macierzyński
Epidemiologia:
Dokładna częstotliwość choroby nie jest znana. W piśmiennictwie niewiele jest danych dotyczących występowania choroby. W północnej Finlandii częstość mutacji A3243G wynosi 16,3: 100 000.
Patogeneza:
Mutacjom mitochondrialnego DNA, które kontrolują łańcuch oddechowy mitochondriów, towarzyszy zakłócenie procesów fosforylacji oksydacyjnej, najważniejszego źródła energii dla procesów metabolicznych w komórce.
Objawy kliniczne
W wieku 40 lat pacjenci z MELAS przyjmowani są do poradni przemijającego napadu niedokrwiennego, a także z padaczką, nawracającymi wymiotami, bólem głowy i osłabieniem mięśni. U tych pacjentów często stwierdza się klinicznie demencję.
Młody wiek i brak czynników ryzyka charakterystycznych dla udaru mózgu sprawiają, że MELAS jest bardziej przemyślany.
Dane laboratoryjne
Kwasica mleczanowa - podwyższony poziom mleczanu i pirogronianu.
Dane wizualizacji
Zmiany w mózgu są podobne do zmian w udarze.
Różnice w stosunku do udaru
1) dotknięte obszary nie pokrywają się z granicami naczyń tętniczych.
2) przy powtarzających się atakach ogniska są wizualizowane w innej lokalizacji.
+ dane kliniczne (młody wiek, brak czynników ryzyka udaru).
CT
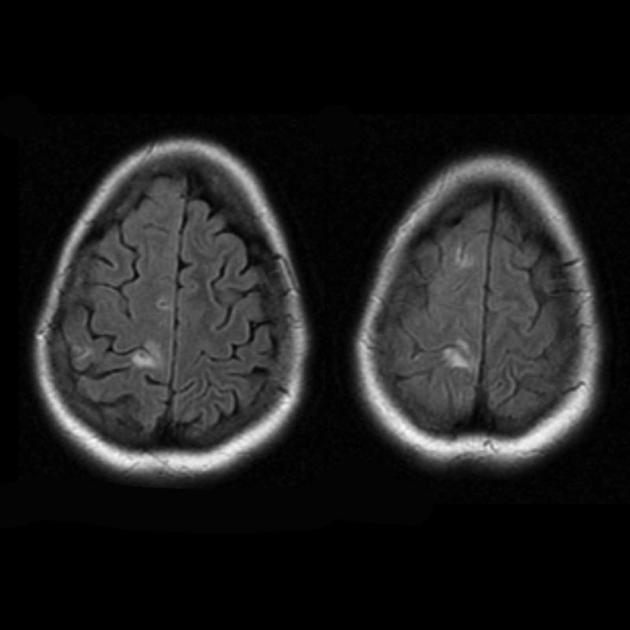
Wiele obszarów hipodensyjnych niezgodnych z łożyskiem naczyniowym.
Zwapnienie jąder podstawy (najczęściej u starszych pacjentów).
Zanik występuje na tle regresji i poprawy klinicznej.
MRI
Ostry zawał
Do różnicowania z udarem stosuje się ADC i DWI (ograniczenie dyfuzji w udarze (obrzęk cytotoksyczny), a w MELAS dyfuzja jest nieznacznie ograniczona lub niezmieniona (obrzęk wazogenny).
Zaangażowanie w patologiczny proces podkorowej istoty białej mózgu.
Pogorszenie wizualizacji klarowności konturów zwojów i wzrost sygnału z nich na obrazach T2-ważonych.
Przewlekły zawał
Zmiany mogą być symetryczne lub asymetryczne.
Zanik ogniskowy występuje na tle regresji i poprawy klinicznej.
Najczęściej atakowane są płaty ciemieniowe, potyliczne i skroniowe mózgu.
spektroskopia MR
Zwiększony poziom mleczanu.



| Miopatia mitochondrialna, encefalomiopatia, kwasica mleczanowa i epizody udaropodobne | |
|---|---|
| Zwapnienie jąder podstawy, zanik móżdżku, zwiększony poziom mleczanu; Obraz TK osoby, u której zdiagnozowano MELAS | |
| Specjalność | neurologia |
genetyka
Biopsja mięśnia osoby, u której zdiagnozowano MELAS, ale bez znanej mutacji. (a) Zmodyfikowany trójkolorowy barwnik Gomori pokazuje trochę postrzępionych czerwonych włókien (strzałki). (b) Barwienie oksydazą cytochromu c, pokazujące włókna typu 1, lekko wybarwione i typu II, włókna ciemne i kilka włókien z nieprawidłowymi zbiorami mitochondrialnymi (strzałki). Należy zauważyć, że włókna ujemne oksydazy cytochromu c są zwykle obserwowane w encefalopatii mitochondrialnej, kwasicy mleczanowej i epizodach udaropodobnych (MELAS). (c) Barwienie dehydrogenazą bursztynianową pokazuje kilka postrzępionych niebieskich włókien i intensywne barwienie w mitochondriach naczyń krwionośnych (strzałka). (d) Mikroskopia elektronowa pokazuje nieprawidłowy zbiór mitochondriów z inkluzjami parakrystalicznymi (strzałki), inkluzjami osmiofilowymi (duże strzałki) i wakuolami mitochondrialnymi (małe strzałki).
MELAS jest spowodowany mutacjami w genach mitochondrialnego DNA.
dehydrogenazy NADH
Mutacje w MT-TL1 powodują ponad 80 procent wszystkich przypadków MELAS. Zmniejszają zdolność mitochondriów do wytwarzania białek, wykorzystywania tlenu i wytwarzania energii. Naukowcy nie ustalili, w jaki sposób zmiany w mitochondrialnym DNA prowadzą do określonych oznak i objawów MELAS. Nadal badają skutki mutacji genów mitochondrialnych w różnych tkankach, zwłaszcza w mózgu.
dziedzictwo
Ten stan jest dziedziczony według wzorca mitochondrialnego, który jest również znany jako dziedziczenie matczyne i heteroplazmia. Ten wzór dziedziczenia odnosi się do genów zawartych w mitochondrialnym DNA. Ponieważ komórki jajowe, ale nie plemniki, dostarczają mitochondriów do rozwijającego się zarodka, tylko samice przechodzą przez warunki mitochondrialne dla swoich dzieci. Zaburzenia mitochondrialne mogą pojawiać się w każdym pokoleniu rodziny i mogą dotyczyć zarówno mężczyzn, jak i kobiet, ale ojcowie nie przekazują mitochondrialnych cech swoich dzieci. W większości przypadków osoby z MELAS dziedziczą zmieniony gen mitochondrialny od matki. Rzadziej zaburzenie wynika z nowej mutacji w genie mitochondrialnym i występuje u osób bez rodzinnej historii MELAS.
diagnostyka
Leczenie / rokowanie
Pacjenci są leczeni w zależności od tego, jakie obszary ciała są dotknięte w danym momencie.
Biuletyn AGIUV, wydanie specjalne, 2013 udk 616.8-007: 616.853.3
początek objawów melas z drgawkami gorączkowymi
(studium przypadku)
następnie. Musabekowa, A.i. Chamzin
Kirgisko-Rosyjski Uniwersytet Słowiański, Wydział Neurologii i Neurochirurgii, Biszkek, Kirgistan
Drgawki gorączkowe (FS) są znane od starożytności. Hipokrates napisał również, że ZF najczęściej występuje u dzieci w pierwszych 7 latach życia, a znacznie rzadziej u dzieci starszych i dorosłych. Ale po raz pierwszy termin „konwulsje gorączkowe” został użyty w 1904 roku przez B. Hochsinge w odniesieniu do napadów drgawkowych, które rozwijają się w dzieciństwie na tle gorączki. Obecnie lepiej mówić o napadach gorączkowych (AF) niż FS, ponieważ w obrazie klinicznym tego stanu można zaobserwować nie tylko napady drgawkowe, ale także niedrgawkowe 2]. Zgodnie z definicją ILAE z 1993 r. migotanie przedsionków to napad występujący u dzieci w wieku powyżej 1 miesiąca, związany z chorobą przebiegającą z gorączką, niespowodowaną infekcją ośrodkowego układu nerwowego; bez wcześniejszych napadów u noworodków i napadów nieprowokowanych oraz niespełniających kryteriów dla innych ostrych napadów objawowych. Według projektu klasyfikacji z 2001 r. AF jest klasyfikowane jako grupa schorzeń, które nie wymagają obowiązkowego rozpoznania padaczki. Dlatego AF definiuje się jako epizod napadów padaczkowych występujących u dzieci w wieku 6 miesięcy i starszych. do 5 lat ze wzrostem temperatury podczas choroby wirusowej lub bakteryjnej, która nie jest związana z neuroinfekcją i zaburzeniami metabolicznymi. Prawdziwe AF należy odróżnić od napadów gorączkowych, które mogą być częścią struktury wielu postaci padaczki, takich jak zespół Draveta. W rzadkich przypadkach AF może być pierwszym objawem choroby mitochondrialnej u dzieci.
Zespół MELAS (mitochondrialna encefalomiopatia z kwasicą mleczanową i epizodami podobnymi do udaru) został po raz pierwszy zidentyfikowany jako odrębna postać nozologiczna przez S. Pavlakisa i in. dopiero w 1984 roku. Choroba należy do grupy chorób mitochondrialnych związanych z mutacjami punktowymi w mitochondrialnym DNA, które skutkują naruszeniem produkcji energii w mitochondrialnym łańcuchu oddechowym. Wiadomo, że mutacje punktowe mogą występować w wielu genach (MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2) i być dziedziczone przez linię matczyną. Częstość występowania zespołu MELAS jest trudna do oszacowania ze względu na różnorodność objawów i związane z tym trudności diagnostyczne. do 2000 roku opublikowano ponad 120 obserwacji choroby. Głównymi objawami w zespole MELAS są: nietolerancja wysiłku, epizody udaropodobne, drgawki, "rozerwanie czerwonych" włókien w biopsjach tkanki mięśniowej, kwasica mleczanowa i początek choroby przed 40 rokiem życia. Zespół MELAS należy odróżnić od innych chorób mitochondrialnych: zespołu Kearnsa-Seyera i MERRF.
Poniżej nasza własna obserwacja pacjenta P., urodzonego w 2003 roku, mieszkającego w Biszkeku. Wiosną 2013 roku do ośrodka kliniki MEBI LTD w Biszkeku zgłosiło się do nas dziecko z dolegliwościami drgawek toniczno-klonicznych w nogach i ramionach trwających do 2 minut, przebiegających z utratą przytomności i rozwijającą się.
tylko na tle wzrostu temperatury ciała powyżej 37 ° C, a także pojawienia się trudności w opanowaniu materiału szkolnego, utraty pamięci, zwiększonego zmęczenia i osłabienia mięśni, niezręczności podczas chodzenia.
Debiut choroby u dziewczynki odnotowano w wieku 6 miesięcy z uogólnionym napadem toniczno-klonicznym trwającym do 1 minuty na tle wzrostu temperatury ciała do 38 ° C, po czym została hospitalizowana w Republice Szpital Chorób Zakaźnych w Biszkeku, gdzie wykluczono neuroinfekcję. Następnie migotanie przedsionków pojawiało się za każdym razem, gdy temperatura ciała przekraczała 37°C. W wieku 1 roku, kontaktując się z Narodowym Centrum Pediatrii i Chirurgii Dziecięcej Republiki Kirgiskiej, wykonano rezonans magnetyczny mózgu, EEG, gdzie nie wykryto patologii, przepisano depakinę w dawce 20 mg/kg/ dzień. Jednak AF trwało nadal podczas przyjmowania leku przeciwdrgawkowego. W wieku 5 lat niezależnie zgłosili się do Republikańskiego Dziecięcego Szpitala Klinicznego (RCCH) w Moskwie, gdzie powtórzono MRI mózgu i monitorowanie wideo-EEG (VEM) snu w ciągu dnia, gdzie ponownie nie wykryto patologii. Lekarze RCCH zdiagnozowali u niego padaczkę kryptogenną i zalecono zwiększenie dawki depakiny do 25 mg/kg/dobę. Skończyłam pierwsze trzy klasy w liceum ogólnokształcącym z klasami „4” i „5”. Od 9 roku życia matka zaczęła zauważać stopniowy wzrost szybkiego zmęczenia dziecka po wysiłku fizycznym, pojawienie się trudności w opanowaniu materiału szkolnego, pojawiło się pytanie o przeniesienie dziecka do specjalistycznej placówki szkolnej dla dzieci z upośledzeniem umysłowym. Migotanie przedsionków nadal przeszkadzało dziecku nawet po 7 latach przyjmowania depakin chrono w dawce 25 mg/kg/dobę.
Z wywiadu życiowego: dziecko z pierwszej ciąży przebiegające na tle łagodnej zatrucia w I trymestrze, miało epizod SARS bez gorączki w okresie 5 miesięcy. Poród w terminie, niezależny w prezentacji głowowej, punktacja Apgar 7/8, CM - 3340 gr., wzrost - 52 cm Wczesny rozwój dziecka odpowiadał normie wiekowej.
W momencie badania w klinice w wieku 10 lat, ze strony nerwów czaszkowych stwierdzono lekkie zbaczanie języka w prawo, zespół miopatyczny w ramionach i nogach w postaci niedociśnienia, łagodne hipotrofie proksymalne partie ramion i nóg ze spadkiem siły mięśniowej do 4 punktów, spadkiem odruchów ścięgnistych, a także lekkim zachwianiem w pozycji Romberga i niezręcznością przy wykonywaniu testów palcowo-nosowych i kolanowo-piętowych, spadek w pamięci krótkotrwałej i uwagi.
Badania dodatkowe: Hb 112 g/l, erytrocyty 3,5*10”12/l, próby wątrobowe, białko całkowite, cukier, kreatynina w normach.
Kontynuacja AF po 5 latach wraz z rozwojem oporności na walproinian, dodanie zespołu miopatycznego i osłabienie funkcji poznawczych pozwoliły na zasugerowanie, że pacjent może mieć patologię mitochondrialną, czyli zespół MELAS, co wymagałoby
przeprowadzenie szeregu dodatkowych badań. Elektroneuromiografia wykonana w NCP i DC wykazała pierwotny typ zmiany mięśniowej w postaci skrócenia czasu trwania potencjału jednostek ruchowych o 30-35% i zmniejszenia ich amplitudy przy normalnej szybkości przewodzenia wzdłuż nerwów obwodowych . Przy powtarzającym się VEM nie wykryto patologii. W laboratorium SVS im. V.M. Savinov miasto Ałmaty oznaczono stężenie depakiny we krwi przed przyjęciem leku - 86,98 ng / ml i 2 godziny po przyjęciu leku - 113,61 ng / ml w tempie 50-100 ng / ml. W klinice Privat w Ałmaty oznaczono zawartość kwasu mlekowego na czczo – 3,1 mmol/l, w ilości do 1,7 mmol/l. Postawiono wstępne rozpoznanie zespołu MELAS, zastąpiono depakinę keppra, wprowadzono koenzym Q10, karnitynę, witaminy z grupy B, witaminę E, zalecono dietę niskowęglowodanową oraz zalecono wykonanie badania genetycznego.
Tak więc w przedstawionym przez nas przypadku klinicznym dziecko miało proste AF, do którego profilaktyki przepisano depakinę z długotrwałym stosowaniem przez kilka lat, pomimo obecności lekooporności. Biorąc pod uwagę brak jednoznacznych dowodów na skuteczność profilaktycznego stosowania leków przeciwdrgawkowych u dzieci z AF, powołanie depakiny w tym przypadku było niewłaściwe. Tak więc, zgodnie z literaturą, długotrwałe stosowanie depakiny i barbituranów znacząco pogarsza przebieg chorób mitochondrialnych, prowadząc niekiedy do progresji procesu patologicznego, co miało miejsce w naszym przypadku klinicznym.
bibliografia:
1. Guzewa VI. Zespoły specjalne (napady sytuacyjne) / V.I. Guzeva // Padaczka i niepadaczkowe stany napadowe u dzieci.-M.: MIA, 2007.- P. 443-457
2. Mukhin K.Yu. Drgawki gorączkowe / A.S. Petrukhin
// Neurologia dzieciństwa. - M.: Medycyna, 2004.-S.664-668.
3. Nikanorova M.Yu., Temin P.A., Kobrinsky B.A. Drgawki gorączkowe / P.A. Temina, M.Yu. Nikanorova // Zespoły padaczki i drgawki u dzieci.- M.: Medycyna, 1999.- P. 169-195.
4. Główne metody leczenia dzieci cierpiących na choroby mitochondrialne: Metodyczne. instrukcje.-M., 2001.
5. Temin P.A. itp. //Nevrol. czasopismo - 1998. nr 2. - s. 43.
6. Yakhno N.N. itp. //Nevrol. czasopismo 1998.- nr 5.- str. 14.
7 Ban S. i in. // Działaj Pathol. Jpn.- 1992.-t. 42.-s. 818.
8. Hirano M., Pavlakis S.G. // J. Clin. Neurol. -1994.-t. 9.-p. 4.
9. Raport Komisji ILAE: słowniczek terminologii opisowej dla semiologii napadowej: raport Grupy Zadaniowej ILAE ds. Klasyfikacji i Terminologii / Padaczka.- 2001.- Cz. 42.-P.1212-1218.
10.ILAE. Wytyczne do badań epidemiologicznych nad padaczką /Epilepsia.- 1993.-Tom.34.-P.592-596.
11. Pavlakis S.G. i in. //Ann. Neurol.-1984. -Tom. 16.-P. 481.
12. Sciacco M. i in. // J. Neurol. -2001. -V 248. -P. 778.
Drgawki gorączkowe mogą być często pierwszym objawem chorób mitochondrialnych u dzieci, co znacznie utrudnia terminową diagnozę i rozpoczęcie patogenetycznego leczenia choroby, a niekiedy powoduje stosowanie leków pogarszających przebieg i rokowanie choroby.
Słowa kluczowe: drgawki, terapia.
Drgawki gorączkowe często mogą być pierwszym objawem choroby mitochondrialnej u dzieci, co znacznie utrudnia terminową diagnozę i wczesne leczenie, a niekiedy powoduje, że stosowanie leków pogarsza przebieg i rokowanie choroby.
Słowa kluczowe: drgawki, terapia.
zł 616.831-005.4
PRZYPADKI Udaru niedokrwiennego jako przejaw encefalopatii mitochondrialnej u młodocianego pacjenta
WIEK
Karbozova K.Z., Łucenko I.L.
Klinika Neurologii z kursem genetyki medycznej Kirgiskiej Państwowej Akademii Medycznej im. I.K. Achunbajewa,
Biszkek, Kirgistan
Częstość występowania udaru mózgu w młodym wieku (do 45 lat) wynosi od 2,5 do 10% wszystkich przypadków udarów naczyniowo-mózgowych i stale rośnie. U młodych pacjentów najczęstszymi przyczynami zaburzeń niedokrwiennych naczyń są: anomalie układu mózgowo-naczyniowego, rozwarstwienie, patologia serca, migrena, wady krzepnięcia, AFLS.
W ciągu ostatnich 5 miesięcy w Klinice Neurologii nr 1 Narodowego Szpitala Ministerstwa Zdrowia Kirgistanu (NHMZKR) leczono 608 pacjentów. Przeanalizowano 46 (7,5) historii przypadków pacjentów z udarem niedokrwiennym mózgu, z których 4 (8,7%) było młodych (poniżej 45 lat według WHO). W tabeli 1 wymieniono podtypy udaru niedokrwiennego.
Przedstawiamy historię chorego hospitalizowanego z wstępnym rozpoznaniem ostrego incydentu naczyniowego mózgu (ACI), u którego prawdziwy charakter choroby można było ustalić jedynie na podstawie obserwacji dynamicznej i specjalnego badania dodatkowego.
Tabela 1.
Podtyp udaru Liczba pacjentów w %
Miażdżyca zakrzepowa 32 69,5
Lakunar 6 13.04
Hemoreologiczny 3 6,5
Cardioembolic 3 6,5
mitochondrialny 1 2,2